Atorvastatin exerts its anti-atherosclerotic effects by targeting the receptor for advanced glycation end products
Bo Feng ,Lei Xu , Hua Wang , Xinfeng Yan , Junli Xue , Fengjing Liu, Ji-Fan Hu
1. Introduction
It is well established that people with diabetes mellitus have a greater risk of cardiovascular morbidity and mortality than their normal counterparts.More than 50% of diabetes-related deaths are associated with macro-vascular complications, especially atherosclerosis. Multiple risk factors are associated with atherosclerosis,including hyperglycemia, hyperlipidemia, and dysregulation of the angiotensin system.
Recent studies show that oxidative stress and advanced glycation also play an important role in the development of diabetic macro-vascular disease. AGE contributes to the progression of atherosclerosis and accelerates oxidative stress by interacting with a specific receptor RAGE on vascular cells .
RAGE, a member of the immunoglobulin superfamily of cell surface molecules, is a multi-ligand receptor on vascular cells that plays a key role in inflammatory processes. Unlike other receptors that are down regulated by increased levels of their ligands, the RAGE-ligand interaction leads to positive feedback activation,which further increases receptor expression . There is increasing evidence that in diabetic patients, activation of the AGE-RAGE pathway plays a central role in the cascade of events that result in accelerated atherosclerotic plaque formation, plaque erosion and fissuring .
In addition to AGE, other cytokines and proinflammatory molecules can also up regulate RAGE expression. For example, MCP-1, a downstream target of the AGE-RAGE signal pathway , is one of these ligands, thus constituting a positive feedback cascade.MCP-1 is an essential chemokine responsible for the recruitment and activation of monocytes to inflammatory lesions in the vasculature, an initial step in the progression towards atherosclerosis .
Recent clinical trials have shown that statins, the competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, reduce cardiovascular events and mortality in diabetics. In animal models, statins exert a beneficial effect on aortic plaque composition, resulting in a decrease in the proportion of advanced plaques and an increase in plaque stability. In addition, statins have many other effects, including anti-inflammation, anti-oxidative stress, and improving endothelial function.These cholesterol-independent effects have been suggested to attenuatemany of the stAGE critical to atherosclerosis (see reviews). However, the specific mechanism of statins on atherosclerosis is still unknown.
We proposed to examine whether atorvastatin exerts part of its inhibitory role on the progression of atherosclerosis by targeting advanced glycation end products. Specifically, we examined the expression of RAGE and its downstream target gene in HUVECs and in Goto Kakisaki (GK) rats. The GK rat was used as a non-obese model of type 2 diabetes. It has been reported that in this model, high-fat feeding markedly impairs glucose-induced insulin secretion in islets and results in a type 2 diabetes phenotype characterized by an increase in fasting hyperglycaemia, plasma triglycerides and cholesterol, impaired early-phase insulin secretion in response to food intake, hepatic insulin resistance, and abnormal glucose metabolism. We were particularly interested in whether in this model,statins alter the AGE-RAGE interaction and suppress their potential target gene RAGE expression in the early stAGE of atherosclerosis, especially in the atherosclerotic aortas far before obvious plaques can be detected.
2. Materials and methods
2.1. Cell culture
RPMI-1640 nutrient medium and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA); VEGF was supplied by Upstate Biotechnology (Lake Placid, NY); Trizol reagent was provided by Invitrogen (Carlsbad, CA); type I collagenase (100 mg) was from Sigma (St. Louis, MO); and the atorvastatin was provided by Honghui pharmaceuticals (Peking, China).
Human umbilical vein endothelial cells (HUVECs)were cultured in our lab and HUVECs at passage 3–4 were used for experiments, and the cells were cultured with serum-free RPMI-1640 medium for 24h before treatment. The HUVECs were randomly assigned to 6 groups as follows: (1) high glucose (30 mmol/L); (2) high AGE (400 μg/ml);(3) high glucose (30 mmol/L) plus atorvastatin (12 mg/L); (4) high AGE (400 μg/ml) with atorvastatin (12 mg/L); (5) control group and (6) high mannitol (30 mmol/L). Cells were cultured at 37 C for 24 h with 5% CO2. After 24 h, the cells were observed and photographed. The culture supernatants of treatment groups were collected for the measurement of glutathione (GSH) and malondialdehyde (MDA), which were used as the anti-oxidative and the oxidative index, respectively.
2.2. Preparation of advanced glycation end products-human serum albumin (AGE-HAS)
Human serum albumin (HSA) and D-glucose were dissolved in PBS (pH 7.2–7.4), and the final concentrations of HSA and D-glucose was 5 g/L and 50 mmol/L, respectively. EDTA was added to a final concentration of 0.5 mmol/L to reduce oxidation. Penicillin (100 U/L) and streptomycin (100 μg/ml) were added to the reaction mixture to prevent bacterial contamination. The reaction mixture was filtered through 0.22 μm filter and then incubated in electrothermal incubation at 37°C for 12 weeks. At the end of the incubation period, the reaction mixture was dialyzed against sterilized PBS (pH 7.2–7.4) to remove the unconjugated glucose; the glucose in the dialyzate was b0.03 mmol/L. The reaction mixture was measured in a fluorospec-trophotometer with an excitation wave of 370 nm, and the maximum absorption peak was measured at 440 nm to verify that the mixture was AGE-HSA. Finally, the AGE-HSA was freeze-dried and stored at 4 °C.
2.3. Animals and diets
Nine male Goto Kakisaki (GK) rats (Scientific research institute,Shanghai,China), weighing approximately 380 g, were randomly divided into two groups: diabetic control group (five rats) and atorvastatin-treated diabetic group (four rats). Five male Wistar rats(Scientific research institute, Shanghai, China), weighing approxi-mately 350g–370 g, were included as the normal control group. Ratsin the atorvastatin-treated group were treated with atorvastatin (Lipitor, Pfizer Ireland Pharmaceuticals), 20 mg/kg/day, through intragastric administration. All rats were fed for 12 weeks with high-fat diet, consisting of 78.5% normal diet, 1% cholesterol, 0.5% bile salt, and 20% lard. All rats were given vitamin D3 (6×105IU/kg)through intragastric administration from the first day to the third day to accelerate atherosclerosis formation. Body weight was measured weekly. Blood samples were drawn from the caudal vein every two weeks until the end of the experiment. Fasting blood glucose levels were measured by glucose oxidase-peroxidase (GOD-POD) kit(Sigma, MO). All animal experiments were conducted according to the protocol approved by the Animal Committee of Animal Center of East Hospital, Tongji University.
2.4. Tissue collection
Rats were sacrificed at the end of 12 weeks and the chest was opened by midline incision. The aortas were rapidly removed and rinsed with ice-cold saline. For each aorta sample, about 1 mm3 was stored in 2.5% glutaral for analysis by transmission electron microscope (TEM). The remaining portions of aorta were frozen by dipping in liquid nitrogen, and then stored at −80 °C.
2.5. mRNA quantitation by real-time PCR
Total RNAwas isolatedwith the trizolmethod and depuratedwith a RNAeasy kit (Invitrogen, CA). RNA was stored at −80 °C until reverse transcription was performed. An aliquot (1 μg) of extracted RNA was reverse-transcribed into the first strand of complementary DNA (cDNA) at 42 °C for 40min, using 100 U/ml reverse-transcriptase (Takara Biochemicals, Shiga, Japan) and 0.1 μM of oligo (dt)-adapter primer(Takara) in a 50ul reaction mixture. Real-time polymerase chain reaction (PCR) was carried out with an ABI Prism 7000 Real-Time PCR system, using the DNA-binding dye SYBER Green I for the detection of PCR products. The reaction mixture (RT-PCR kit, Takara) contained 12.5 μl Premix Ex Tag, 2.5 μl SYBER Green I, custom-synthesized primers, ROX reference dye, cDNA (equivalent to 20 ng total RNA) to give a final reaction volume of 25 μl. Primers were as follows: β-actin: sense 5′,TCTGTGTGGATTGGTGGCTCT 3′,antisense5′ AGAAG-CATTTGCGGTGCAC 3′; RAGE: sense 5′CCTGTGGCGAAAACGACAA3′,antisense5′TCTGGCATTTCCGCTTCCT3′;MCP-1:sense 5′CAGATGCAGT-TAATGCCCCA3′,antisense5′CCTGCTGCTGGTGATTCTCTT3′.The PCR settings were as follows: initial denaturation of 15min at 70 °C,followed by 40 cycle of amplification for 15 s at 95 °C and 1 min at 59 °C, with subsequent melting curve analysis increasing the temperature from 60 to 95 °C. In order to quantify RAGE and MCP-1 gene expression, the RAGE and MCP-1 mRNA level was normalized by internal β-actin mRNA.
2.6. Transmission electron microscopy (TEM) test
After fixation in glutaral, aortas were put into 1% osmic acid, dehydrated with acetone, embeddedwith Epon812, slicedwith LKB-V microtome, double stained with lead and uranium, and observed under an H-600A transmission electron microscope.
2.7. Data analysis
All data were expressed as mean±SD. Kruskal–Wallis one-way analysis of variance was used to assess the differences between inner-groups and inter-groups using SPSS 11.5 software. Values of Pb<0.05 were considered to be statistically significant.

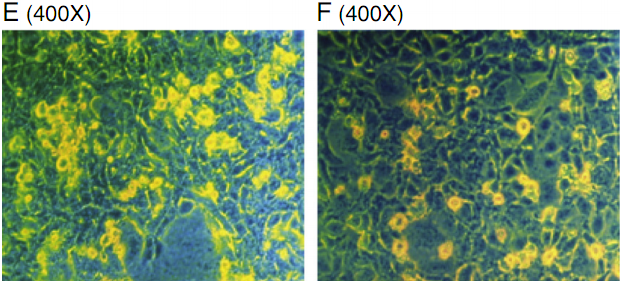
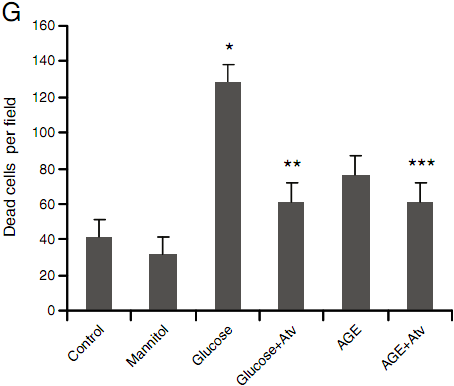
Fig. 1. Cell growth and morphologic changes of HUVEC treated with high glucose and AGE. HUVE cells were treated for 24 h with A: PBS control; B: high mannitol control(30mmol/L);C:high glucose (30 mmol/L); D: high glucose+atorvastatin (12 mg/L); E: high AGE (400 μg/ml); F: high AGE with atorvastatin (12 mg/L); G: Cell death in each treatment group(quantitated as dead cells/microscopic field). Data are expressed as mean±SD in each group. * compared with the control group, Pb<0.05; ** compared with the high glucose group, Pb<0.05; *** compared with the AGE group, P=0.5. Photos were taken 24 h after treatment. (Dead cells are shown in yellow).
3. Results
3.1. Atorvastatin-mediated reduction of abnormal HUVEC growth from high glucose and AGE
Vascular disease caused by diabetes mellitus is related to high serum glucose and AGE levels. In the in vitro model, we incubated HUVECs with high glucose and AGE (Fig. 1). Exposure to high glucose and AGE resulted in a reduction in cell growth, primarily due to prominent cell death (Figs. 1C, E). However, treatment with atorvastatin reduced cell death caused by high glucose (Fig. 1D) and AGE (Fig. 1F). As the treatment control, highmannitol did not alter cell morphology and cell growth (Fig. 1B), indicating that hyperosmosis has no significant impact on the HUVECs. For comparison, cell death in each treatment group was quantitated and summarized in Fig. 1G.
3.2. Biochemical measurement of oxidative stress
AGE accelerates oxidative stress by interacting with a specific receptor RAGE on vascular cells. To determine the involvement of oxidative stress in cell death of HUVECs, we measured GSH (indicator of redox status) and MDA (indicator of lipid peroxidation induced by oxygen free radicals) in cell supernatants (Fig. 2). The mannitol treatment did not alter GSH levels as compared with the control group. However, there was significant decrement in GSH after exposure of the cells to high glucose or AGE, indicating an increased oxidative stress of the treated cells. Treatment of the cells with atorvastatin significantly released the oxidative stress by restoring the levels of GSH (Fig. 2A) (Pb<0.01).
Similarly, incubation of cells with high glucose and AGE caused a dramatic increase in MDA production. Atorvastatin suppressed the production of lipid peroxidation from oxygen free radicals following the treatment of high glucose and AGE (Fig. 2B) (Pb<0.01).
3.3. Atorvastatin targets RAGE expression in HUVECs
To further explore the role of atorvastatin in protecting HUVECs from cell death, we measured the expression of its target genes. We initially focused on the RAGE gene. After incubation with high glucose and AGE, HUVECswere collected and used for quantitation of RAGE by real-time PCR. As seen in Fig. 3, both high glucose and AGE-induced up regulation of RAGE mRNA in HUVECs. However, treatment of cells with atorvastatin led to the down regulation of RAGE to the basal level seen in both PBS and mannitol control cells.
3.4. Downregulation of RAGE and its downstream target MCP-1 by atorvastatin in GK rats
Given the beneficial effect of atorvastatin in HUVECs, we further examined its therapeutic effect in a diabetic GK rat model. After being fed a high-fat diet for 12 weeks, blood glucose levels were significantly increased in diabetic group (n=5, 12.7 mmol/L±4.2 mmol/L) as compared with that in the normal control group (n=5, 5.9±0.2 mmol/L) (Pb<0.01) (Fig. S1). No significant effect on plasma glucose was observed in atorvastatin-treated diabetic group.
To further examine the role of atorvastatin in this GK ratmodel,we used RT-PCR to quantitate the expression of RAGE in the aorta. As compared with the normal control group, expression of RAGE mRNA was significantly increased in the diabetic control group (Fig. 4A) (P<0.01). In the atorvastatin-treated diabetic group, however, RAGE mRNA levels were significantly downregulated to a level comparable to those observed in the normal control group.
In addition to AGE, there is a potential link between inflammation and insulin resistance in obese mice. Thus, we measured the expression of MCP-1, which is a downstream target of the AGE-RAGE pathway, and also plays an important role in the recruitment and activation of monocytes in rat aorta.In this study, we observed a significant correlation between the abundance of RAGE mRNA and
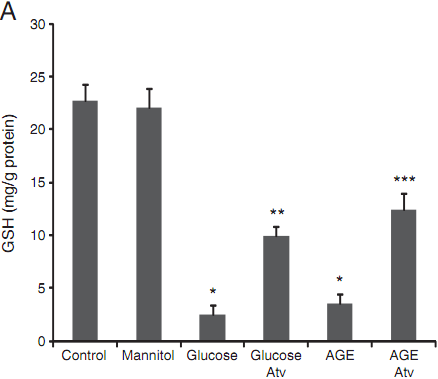
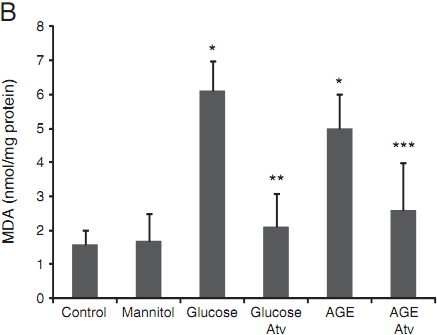
Fig. 2. Measurement of oxidative indicators GSH (A) and MDA (B) in the collected supernatants of treated HUVEC. Data are expressed as mean±SD in each group.* compared with the control group, Pb<0.01; ** compared with the high glucose group,Pb<0.01; *** compared with the AGE group, Pb<0.05.
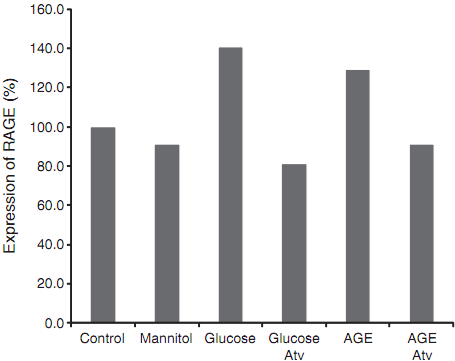
Fig. 3. Expression of the receptor for the advanced glycation end products (RAGE) in HUVEC received varied treatments. The abundance of RAGE mRNA was measured by real-time qPCR, standardized over internal control β-actin mRNA, and expressed using the PBS control as 100%.
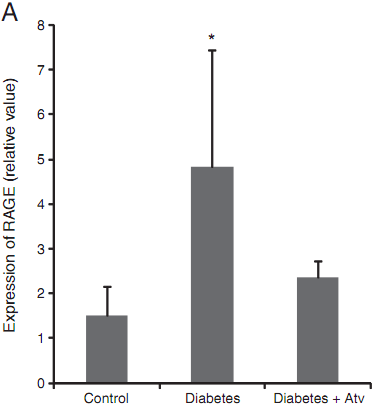
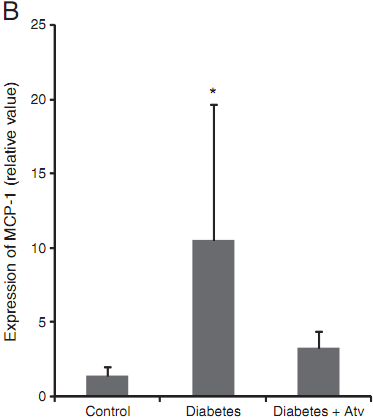
Fig. 4. Atorvastatin downregulated RAGE (A) and MCP-1 (B) in diabetic GK rats.Diabetic control group: five GK rats; atorvastatin-treated diabetic group: four GK rats;and normal control group: five male Wistar rats. Data are expressed as mean±SD in each group. * compared with the control group, Pb<0.05.
MCP-1 mRNA (r=0.482, P=0.031), supporting a possible link between these two molecules in the amplification of inflammatory responses. As seen in Fig. 4B, MCP-1 was significantly up regulated in the aorta of the diabeticmice. Treatment of atorvastatin reduced MCP-1 production, supporting a second pathway by which the statin protects mice from high-fat diet-induced diabetes.
3.5. Morphologic changes of TEM
We also used transmission electron microscopy (TEM) to examine structural alteration in the aortas of GK rats fed a high-fat diet. Early pathological changes of atherosclerosis were observed in the aortas of diabetic rats (Fig.5), such as proliferation of smooth muscle cells(SMC), lipid droplet in the cytoplasm of SMC, proliferation of fiber between SMC, lipid droplet in the basal membrane of aortas,endometrial thickening. No significant improvement in the pathological changes was noticed by atorvastatin treatment in this model.
4. Discussion
As an inhibitor of HMG-CoA reductase, atorvastatin is used extensively to lower serum cholesterol levels. Reduction of total and low-density lipoprotein cholesterol by atorvastatin has been shown to translate into the reduced risk of long-term morbidity and mortality related to coronary artery disease (see reviews). Depending on the model used, animals fed a high-fat diet usually develop hyperlipidemia. Atorvastatin therapy is very effective in lowering serum lipid levels in these animals, including cholesterol and triglyceride . For example, Hamed et al.reported that male Sprague–Dawley rats fed a high cholesterol diet for 8 weeks increased serum cholesterol by 257% (pb<0.001) as compared to the control group fed standard diet. Low dose of atorvastatin induced reduction in cholesterol level by 40% (Pb<0.001). Combined treatment of atorvastatin with other reagents, like garlic extracts, induced even more pronounced hypolipidemic effect.
The efficacy of statin therapy for reducing mortality and morbidity of cardiovascular disease (CVD) in patients with diabetes is now well established. However, statins may be able to reduce cardiovascular events by mechanisms other than its ability to lower the level of serum lipids, i.e. the "pleiotropic" actions. This concept has been further proven by experimental and clinical studies that have shown that statins exhibit a variety of nonlipid effects, including immuno-modulation, anti-proliferation, and anti-thrombosis, improvement of endothelial dysfunction, increased nitric oxide bioavailability, antioxidant properties, inhibition of inflammatory responses,and stabilization of atherosclerotic plaques (See reviews).RAGE activation is known to be associated with activation of the NF-kappaB pathway. The RAGE gene promoter contains three putativeNF-kappaB-like binding sites that respond to the inflammatory stimuli. On the other hand, the activation of the AGE/RAGE also results in generation of intracellular oxidative stress and subsequent activation of the redox-sensitive transcription factor NF-kappaB. The activation of the NF-kappaB pathway by RAGE may further amplify RAGE expression in inflammatory cells, constituting a positive control cascade between oxidant stress and the altered AGE-RAGE system. It has been suggested that statins may reduce AGE accumulation and reduce activation of NADPH oxidase. We also found that atorvastatin treatment improved the oxidative stress index and attenuated inflammation, and reduced cell death of human umbilical vein endothelial cells following exposure to advanced glycation end products. The data in this study, however, suggest that atorvastatin exerts its therapeutic role at least in part by targeting the RAGE activation.
Using the high-fat feeding GK rats as a model, we also show that atorvastatin down regulates RAGE in the very early stAGE of atherosclerosis, before its therapeutic improvement of the atherosclerotic lesions can be detected histologically. Interestingly, we also found that this gene downregulation on the AGE-RAGE system was not accompanied by the altered serum glucose (Fig. S1), indicating a glucose-independent mechanism. Thus, our data demonstrate that down regulation of the RAGE gene may be a novel "pleiotropic"mechanism of atorvastatin in reducing the risk of cardiovascular diseases.
However, it is still unclear how atorvastatin modifies the expression of genes involved in the RAGE pathway. Does atorvastatin down-regulate the RAGE gene directly by binding and interferingwith its promoter activity, or indirectly by modifying the epigenotype around the RAGE gene promoter, like DNA methylation and/or histone acetylation and methylation? Atorvastatin has been reported to affect expression of many other genes involved in pathogenesis of cardiovascular diseases, like peroxisome proliferator activated receptor-γ (PPAR-γ), lysyl oxidase, monocyte chemoattractant protein-1 (MCP-1), interleukin-8 (IL-8), plasminogen activator inhibitor-1 (PAI-1) , and nitric oxide synthase (iNOS).
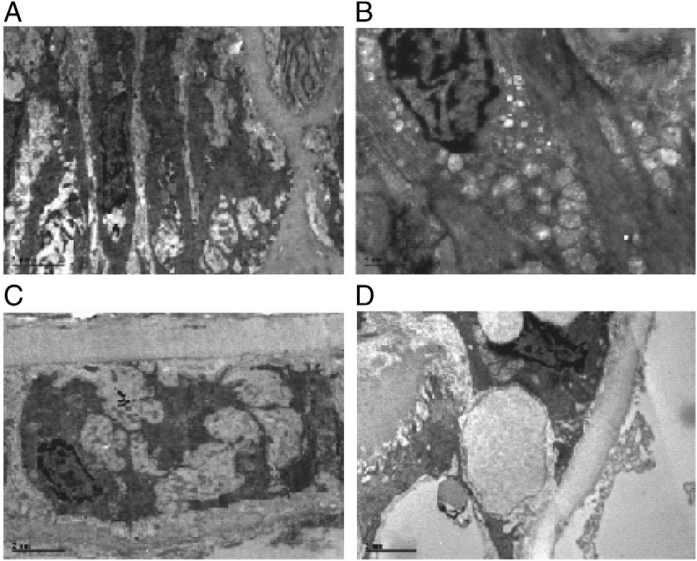

Fig. 5. Morphological changes of aortas as observed by transmission electron microscopy (TEM).
A: Proliferation of smooth muscle cells (SMC) in aortas of diabetic rats (×6000);B: Lipid droplet in the cytoplasm of SMC in aortas of diabetic rats (×8200); C: proliferation of fiber between SMC in aortas of diabetic rats (×8200); D: lipid droplet in the basal membrane of aortas of diabetic rats (×6000); E: SMC in aortas of normal control group (×8200); F: Endothelial Cells in aortas of normal control group (×4200).
It was recently found that atorvastatin was able to rebalance the gene expression profile (165 elevated and 281 suppressed genes)induced by the atherogenic diet . Thus, it is highly possible that atorvastatin modifies an array of genes through an epigenetic mechanism. Further studies are merited to examine the mechanism underlying gene regulation using epigenetic techniques, like DNA methylation sodium bisulfite sequencing and chromatin immunoprecipitation (ChIP).
AGE-RAGE interaction can be blocked by truncated soluble isoforms of the receptor, referred to as total soluble RAGE (sRAGE), derived from either the products of endogenous splice variants (endogenous secretary receptor or esRAGE), or the proteolytically cleaved forms shed into the bloodstream upon digestion of extracellular metalloproteinases. These truncated sRAGE can modulate AGE-mediated pathogenesis and immune modulation, probably by acting as an endogenous decoy protecting factor to exert their anti-atherogenic effects. Indeed, hypercholesterolemic subjects have significantly lower circulating sRAGE levels than age-matched control subjects.Interestingly, treatment of atorvastatin restores the sRAGE levels near to normal values. Tam et al. demonstrated that atorvastatin increased serum sRAGE and esRAGE in type II diabetic patients by inducing their production in macrophAGE in a time- and dose-dependent manner. However, it remains to be investigated whether atorvastatin increases sRAGE production at the post-transcription level or at the level of gene transcription as in the case of RAGE observed in this study.
Most previous studies have primarily focused on the effect of atorvastatin on RAGE expression in atherosclerotic plaque and on the plaque’s instability. In our study, higher expression of RAGEwas found in aortas of diabetic group rats compared with normal control group even in the very early stage of atherosclerosis. Our study demonstrates that diabetic animals demonstrated a significant increment in RAGE expression even before obvious plagueswere detected in the aortas. Still, RAGE expression inGK ratswas significantly reduced by atorvastatin independent of glycemic control. Early expression of RAGE could be caused by the high expression of the main ligands such as inflammatory factors in diabetic aortas.However, the reduction of RAGE by atorvastatin has not fully translated into a significant improvement in early pathological changes. Further studies are needed to examine the long-term effects of down-regulating AGE/RAGE in this rat model.
Clinical evidence has shown that atorvastatin therapy reduces the risk of cardiovascular disease by decreasing serum total cholesterol and low-density lipoprotein cholesterol. However, due to the limit of blood samples collected from the caudal vein, we could not determine serum cholesterol and triglyceride in our GK rats fed a high-fat diet. As a result, we were unable to evaluate the lipid-lowering effect of atorvastatin in the present study that focuses on the RAGE pathway. This limitation will be addressed in our future studies.
There is a growing body of evidence that engagement of RAGE with AGE elicits oxidative stress generation and subsequently evokes inflammatory responses in various types of cells, thus actively participating in the development and progression of atherosclerosis. Therefore, RAGE may play a pro-atherogenic role in diabetic arteries. Okamoto et al. reported that Cerivastatin completely prevented the AGE-induced increase angiogenesis by inhibiting the AGE-induced transcriptional activation in NFKB signaling pathways. Jinnouchi et al. for the first time demonstrated that atorvastatin decreased serum levels of AGE in hypercholesterolaemic type 2 diabetic patients without any cardiovascular disease. In this study, we also demonstrated that atorvastatin suppressed the AGE/RAGE pathway by a gene targetingmechanism. Thus, downregulation of RAGE expression or blockade of RAGE downstream signaling may be a promising target for therapeutic intervention in diabetic Atherosclerosis.
In addition to AGE, many RAGE downstream molecules, like cytokines and proinflammatory products, also upregulate RAGE expression, thus forming a positive feedback cascade. For example, MCP-1 is an AGE-RAGE downstream target. It also acts as one of the important ligands for RAGE and stimulates its expression. In this study, we found that MCP-1 is downregulated in HUVECs by the atorvastatin treatment. Thus, atorvastatin may reduce the activation of the AGE/RAGE pathway partially by breaking the feedback of inflammatory factor MCP-1. Thus, atorvastatin may benefit diabetic patients by blocking the AGE-RAGE signal pathway at multiple levels. RAGE downregulation in diabetic aortas treated with atorvastatinmay establish a negative auto-inhibitory autocrine and paracrine feedback loop, thus delaying the formation and development of atherosclerosis.
In conclusion, this study proposes an etiologic hypothesis regarding statin therapy and atherogenesis in diabetics by demonstrating the inhibition of the functional AGE/RAGE axis after atorvastatin therapy. These findings are also potentially important from a practical standpoint, because they raise the interesting possibility that modifi-cation of RAGE signaling by statins might provide a novel form of therapy for preventing formation and development of atherosclerosis in diabetic patients.
Supplementarymaterials related to this article can be found online at doi:10.1016/j.bbadis.2011.05.007.
Acknowledgments
We thank Amy Truong for editing the manuscript. This study was supported by the Basic Research Program of Shanghai (08JC1419900)to B.F.; NIH grant (1R43 CA103553-01), The Department of Defense Grant (W81XWH-04-1-0597), and California Institute of Regenerative Medicine (CIRM) grant (RT2-01942) to J.F.H.
Biochimica et Biophysica Acta 1812 (2011) 1130–1137